1) Introduction
At OpenADMET, we have the mission to make therapeutic development more reliable, affordable, and effective by building open models and datasets for ADMET properties. To deliver on this goal, we are chemically mapping the “Avoidome,” consisting of the enzymes, transporters, and receptors treated as “antitargets” in medicinal chemistry and pharmacology 1. By uncovering and elucidating the molecular structures that drive activity against antitargets, the next generation of safe and effective medicines will be developed at a fraction of the traditional time and cost.
However, chemical space is vast. Common estimates place the number of possible distinct small organic molecules at around 10^60 2. For context, there are approximately 10^80 nucleons in the observable universe 3. Moreover, many antitargets—particularly PXR (pregnane X receptor)—are known to be promiscuous, binding to a wide range of xenobiotic small molecules to fulfill their role in sensing and regulating metabolism 4. In fact, PXR is so promiscuous that it is often jokingly referred to as the “promiscuous xenobiotic receptor” 5, 6.
In order to deeply understand the precise chemical moieties that drive a small molecule’s activity against an antitarget, developing an approach that allows access to broad swaths of chemical space and extends beyond commercially-available molecules and FDA-approved medicines is key. Screening conventional chemical decks can serve as an insightful medicinal chemistry starting point, but generating the data to subsequently map the structure-activity relationship (SAR) landscape surrounding a target—or in this case, an antitarget—necessitates the synthesis and testing of novel compounds and analogs at scale to comprehensively explore activity cliffs, selectivity, and potency-driving structural motifs.
At Octant, we routinely synthesize and screen tens of thousands of molecules on a weekly basis for our drug programs using direct-to-biology (D2B) approaches. D2B enables greater speed, lower cost, and broader coverage of chemical space by assaying reaction mixtures directly without purification. At Octant, this concept is put into practice through the integration of high-throughput chemistry (HTChem) with high-throughput screening (HTS), where nanoscale chemical synthesis enables the generation of large compound libraries that can be screened at scale.
Fundamental to a D2B workflow is the screening of crude reaction mixtures 7. While efficient for discovery, screening crude mixtures poses a challenge when using D2B data for machine-learning model training. This is due to the confounding characteristics of crude reaction mixtures, including unidentified side-product formation, potentially biologically active reagents and byproducts, and, notably, variable synthesis yields of the intended product that obscure its true concentration and assay readout. In drug discovery, these challenges can be addressed by larger-quantity resynthesis and purification of select compounds to confirm potency and on-target binding. However, this approach does not easily scale to the size and breadth needed for machine-learning datasets, where training useful regression models generally requires large numbers of quantitative data points. At Octant, we have found that by pairing the appropriate analytical infrastructure with cell-based D2B screening, reliable high-throughput potency data for newly-synthesized compounds can be obtained.
In this post, we will describe the development of a method for yield-based potency correction via charged aerosol detection (CAD) to harness the scale of a high-throughput D2B platform while generating data that reflects the fidelity of pure compound screening.
2) Generating Data for OpenADMET
OpenADMET’s PXR induction challenge is currently underway! For this challenge, Octant developed an in-house PXR agonism, cell-based reporter assay from which a dose-response curve (DRC) and potency metric—EC50 (half-maximal effective concentration)—can be determined for small molecules. The need to map PXR’s small-molecule activation landscape is underscored by the fact that reliable, publicly-available EC50 data remain notably sparse relative to PXR’s significance in drug discovery. Specifically, there are only ≈800 high-quality PXR EC50 data points available in ChEMBL (for 657 unique compounds), which were compiled from 148 different papers in which assay conditions were not standardized. In OpenADMET’s first step to extensively explore the activator landscape for PXR, over 11,000 diverse commercially-available compounds and FDA-approved medicines were screened with our PXR assay (Fig. 1A). To explore PXR chemical space further and iterate on the findings from this dataset, HTChem can be employed to build novel chemical libraries by decoupling chemical bonds—such as an amide—in PXR-active scaffolds (Fig. 1B). As will be described in greater detail in Section 6, Octant pursued this strategy by synthesizing and screening a total of 3,181 molecules, including 3,168 compounds synthesized using high-throughput approaches and additional singleton compounds for method validation. From this work, 565 unique compounds were screened with 9-point DRCs, and 454 novel EC50s were generated (and released with this blog post) from molecules that passed both synthesis and screening QC. Over half of these compounds had pEC50 values greater than 5, and the average and median pEC50 were 4.9 and 5.2, respectively.
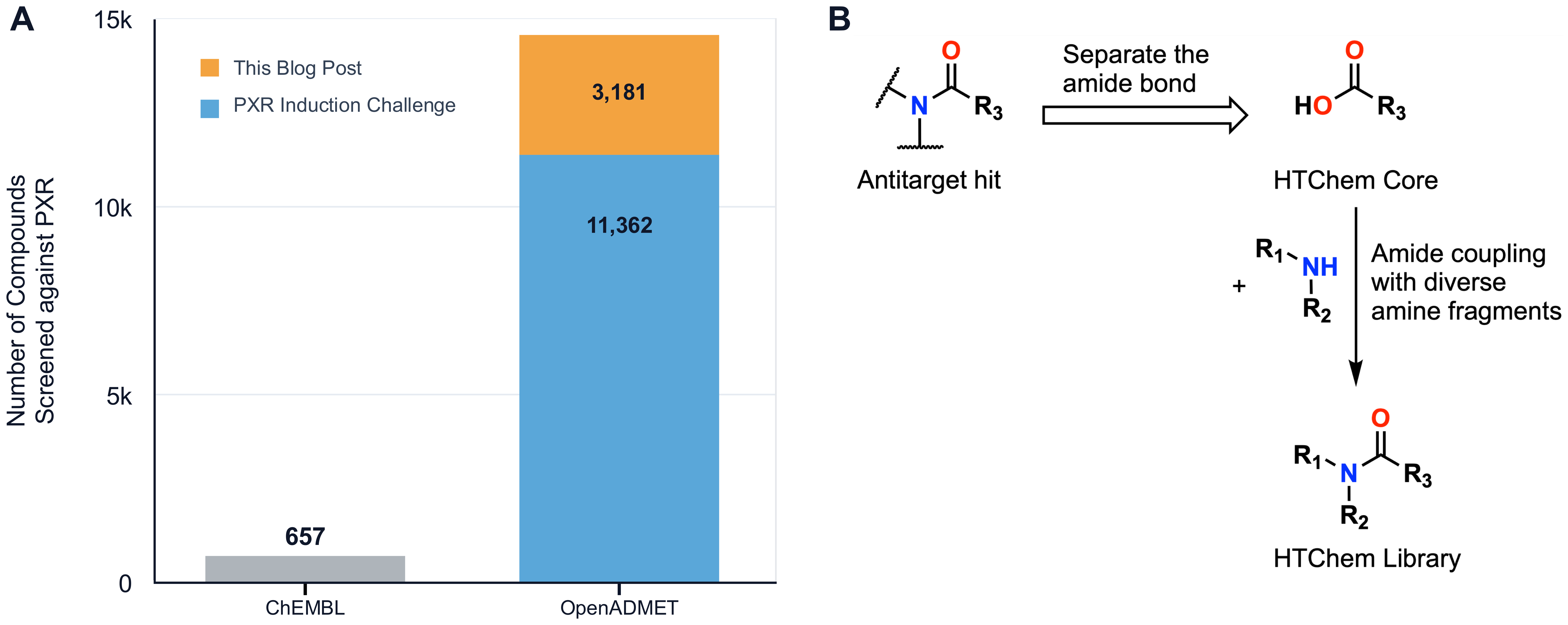
Figure 1:
A. As of May 2026, there are 657 small molecules with PXR activity data in the ChEMBL database. This number is contrasted with the number of compounds that OpenADMET has screened against PXR.
B. A method to design a “core” for HTChem is to disconnect a chemical bond from a known active compound. Then, a novel library of analogs to the reference molecule is synthesized by reacting an amenable fragment set with the core molecule.
HTChem library size can be scaled up (to over 10k analogs from a single core) or refined for a more targeted approach to exploring SAR. However, regardless of library size, HTChem reaction yields can vary significantly from compound to compound, even after pre-synthesis optimization of reaction conditions. Thus, in order to more confidently assign a potency value to an HTChem molecule, incorporation of reaction yield into the calculation of the EC50 is crucial so that potency is determined from the actual concentration of the active species. This process of EC50 “correction” is done by retroactively adjusting the crude, uncorrected EC50 in a manner proportional to the reaction yield (Eq. 1). Equivalently, the correction can be understood as adjusting the nominal concentration of a reaction initially assumed to have had 100% yield by a factor corresponding to the observed yield (Eq. 2). Conceptually, this can be visualized by adjusting the x-axis of a dose-response curve based upon the true concentration of the HTChem molecule. This process of EC50 correction, and the subsequent movement of a data point when comparing a crude HTChem compound’s pEC50 to its pure-version pEC50, is animated below (Fig. 2).
\[\text{Eq. 1} \qquad \text{Corrected EC}_{50}\text{ (M)} = \text{Crude EC}_{50}\text{ (M)} \times \text{Yield (\%)}\]
\[\text{Eq. 2} \qquad \text{True Concentration (M)} = \text{Nominal Crude Concentration (M)} \times \text{Yield (\%)}\]
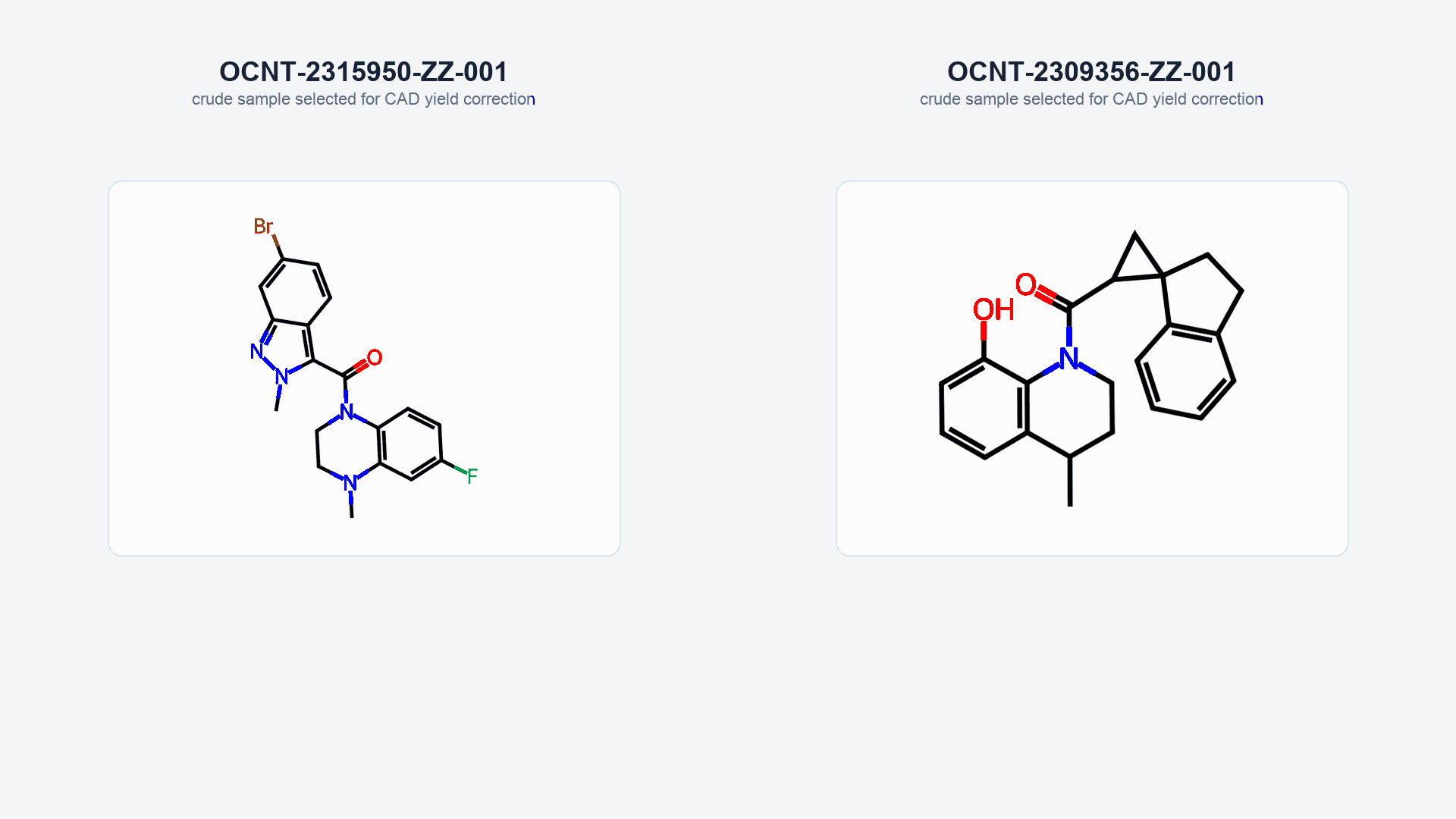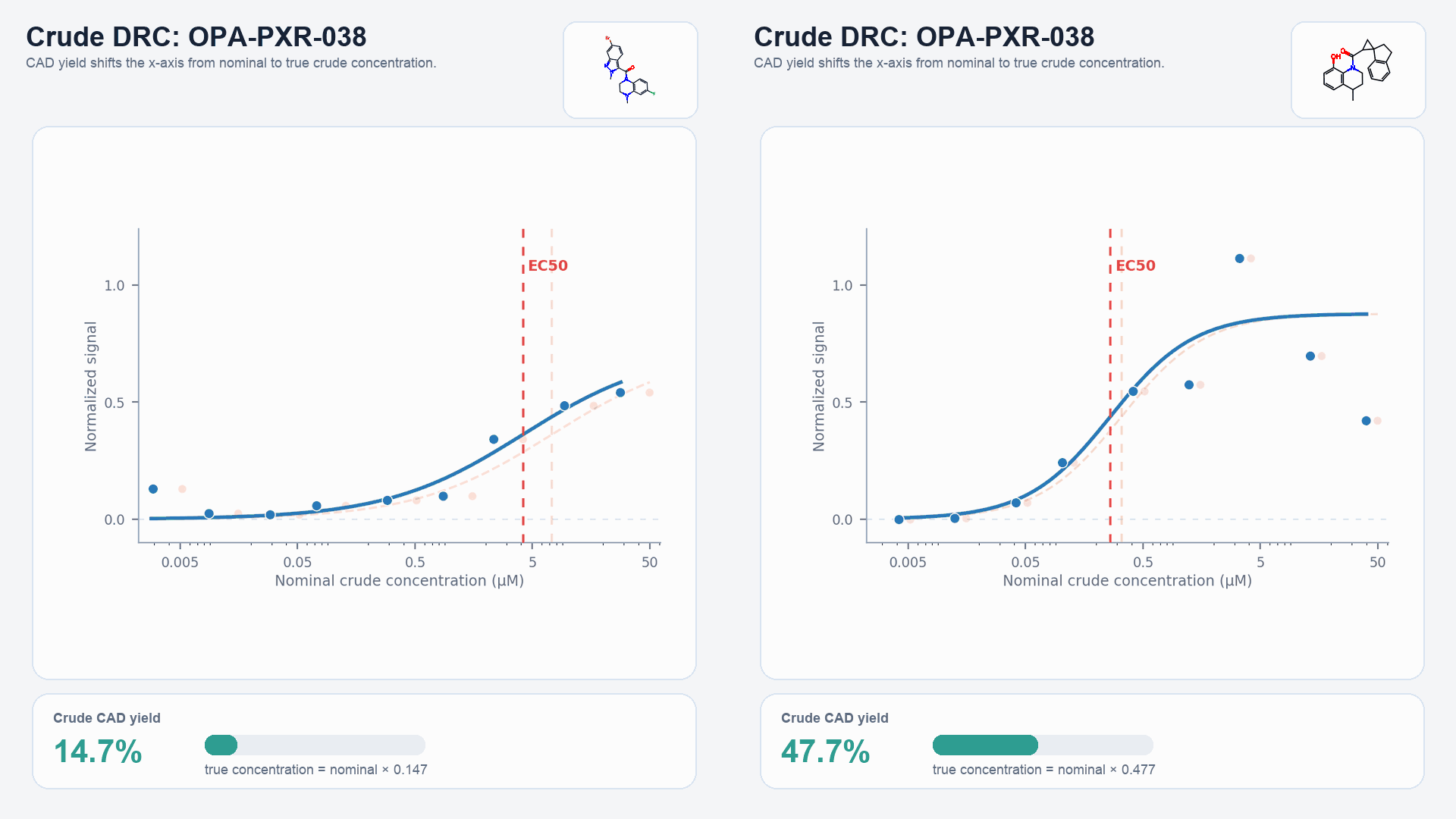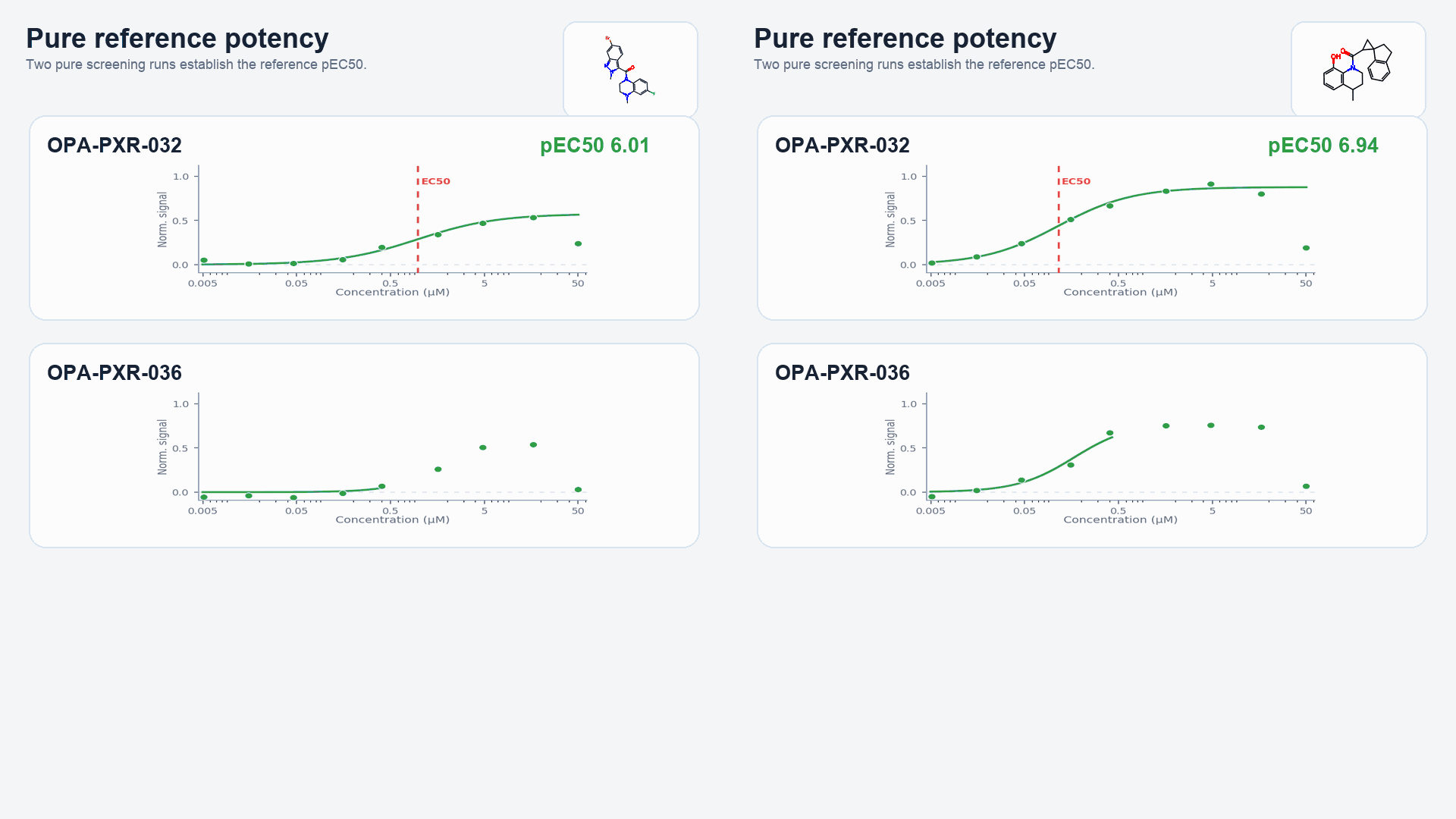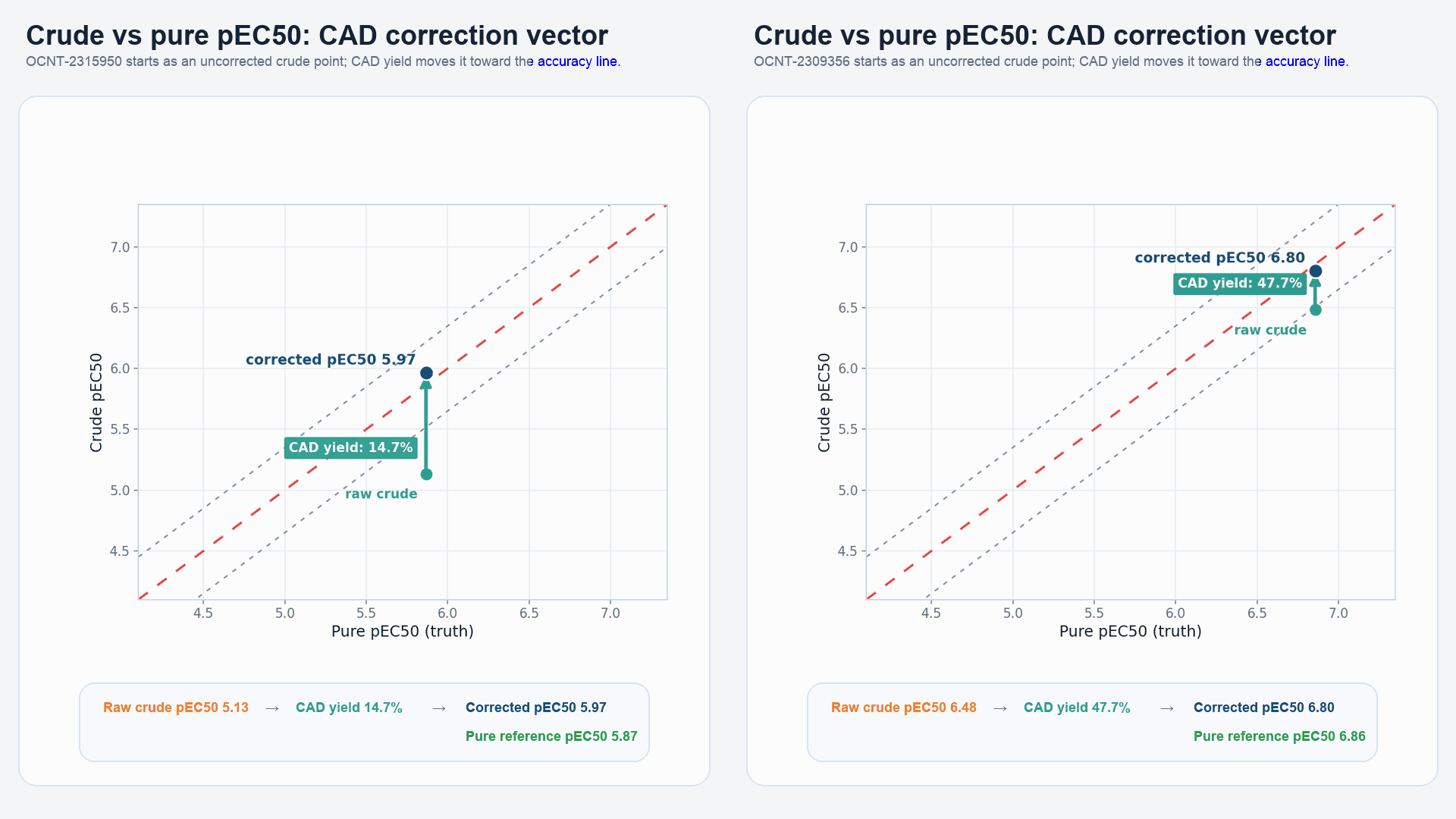
Figure 2: The process of EC50 correction, along with HTChem-to-pure comparisons, is shown with two example compounds: OCNT-2315950 and OCNT-2309356.
- Scene 1: Dose-response curves and CAD-based yields were collected for the crude HTChem compounds.
- Scene 2: The DRC is shifted proportionally to the yields of the crude HTChem reactions.
- Scene 3: Pure versions of the same HTChem compounds were screened in duplicate, and EC50s derived from the DRCs were averaged.
- Scene 4: A plot comparing crude pEC50 (y-axis) and pure pEC50 (x-axis) first depicts the original correlation without yield-based correction, and then—through an arrow animation—shows the movement of the data point on the plot following EC50 correction.
To explore PXR chemical space with HTChem, we selected two of our most potent PXR activators and treated them as two distinct chemical series, much as a medicinal chemist would for a traditional target. The two selected reference compounds showed a preliminary sign of SAR with chemically-similar analogs in the 11k screening deck. By disconnecting their amide bonds, we designed two carboxylic acid HTChem cores to react with a diversity-downselected set of 1,536 fragments bearing a complementary amine functional group. Through this “core-and-fragment” approach, we synthesized two HTChem libraries derived from known PXR-inducing compounds, allowing us to systematically examine how local structural changes influence PXR activation. Additionally, because the reference compounds have been co-crystallized with PXR by the OpenADMET team at UCSF, the binding pocket and fragment-facing orientation of novel hits identified from these HTChem libraries can be analyzed and inferred (Video 1).
Video 1: Exploring PXR Chemical Space
However, the screening of crude D2B libraries poses two intrinsic questions:
Was the intended molecule synthesized? And if so, what was the compound’s yield (and thus true concentration) that determined an EC50 value?
Was activity in an assay driven by the desired product, or by any impurities or side products?
Without answers to these questions, one may argue that the data generated from D2B are not quantitative enough for model training. To close this gap, we undertook a combination of analytical and benchtop chemistry method development.
3) Development of a UHPLC-CAD-MS Method for the Characterization and Quantification of Crude, High-Throughput Chemical Reaction Mixtures
To support meaningful pEC50 interpretation from crude D2B screens, where apparent potency depends directly on true product concentration, we needed an analytical readout that is both molecule-agnostic and capable of converting chromatographic signal directly into product mass. Within the pharmaceutical industry, CAD has been employed for precisely this purpose 8, eliminating the need to purchase or synthesize analytical reference standards for every novel compound produced (an approach that would not scale with HTChem). CAD delivers “near-universal detection” 9 by evaporating the chromatographic mobile phase and leaving behind non-volatile particles, which are then charged and quantified by measuring the resulting current. The signal scales with particle mass largely independent of analyte structure, producing peak areas that reflect molecule abundance far more closely than other analytical techniques such as mass spectrometry, where signal intensity is strongly influenced by ionization efficiency. Prior to the CAD integration, Octant’s analytical workflow had been LC-MS-based, which was well-matched to HTChem synthesis throughput, but unable to deliver standard-free quantitative measurement of reaction outcomes.
At Octant, we have developed a UHPLC-CAD-MS method that enables standard-free absolute quantification of crude reaction mixtures from the HTChem platform, allowing us to characterize reaction yields and subsequently correct EC50s. The method reduces to a single universal calibration relationship between on-column mass (in nanograms) and CAD peak area (in picoampere-minutes).
\[\text{Eq. 3} \qquad \text{Mass (ng)} = 12.5 \times \text{CAD peak area (pA·min)}\]
The 12.5 ng/(pA·min) conversion constant applies to any analyte without prior knowledge of its structure or response factor. Across a calibration set of 300+ structurally-diverse compounds, the inter-analyte coefficient of variation was 26.79% at a standard injection mass of 250 ng and 31.74% with respect to overall slope. The lower limit of quantification (LLOQ) is 1.0 pA·min, which was derived from a 10:1 signal-to-noise ratio (a standard analytical chemistry benchmark) 10. These quantities allow us to quantify uncertainty in the yield estimate, which can then be propagated together with the error from the DRC fit to determine the uncertainty in the final corrected EC50.
To deploy CAD in production, we integrated a Thermo Scientific Corona Veo RS instrument into a UHPLC-CAD-MS configuration in which post-column flow is split between the CAD and a Thermo TSQ Quantis triple quadrupole mass spectrometer (Fig. 3). The CAD provides the quantitative readout, and the MS handles peak identification through alignment of retention times with CAD-detected species. This division of labor is fundamental to the method: the CAD tells us “how much,” and the MS tells us “what.” An inverse gradient arising from a second pump—shown at the bottom of the LC stack in Figure 3—mixes with the effluent post-column to maintain constant total organic composition entering the CAD, mitigating response variability that results from the CAD’s intrinsic sensitivity to mobile phase composition 11, 12.
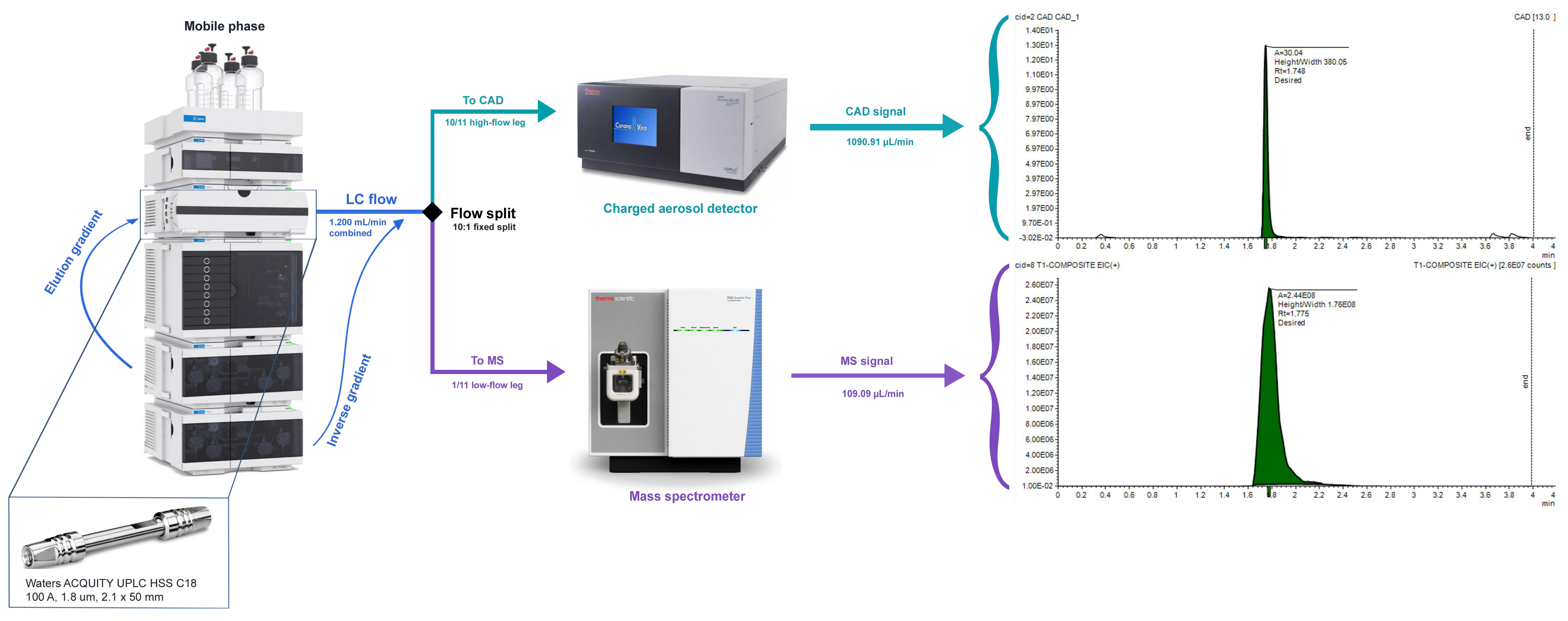
Figure 3: The UHPLC-CAD-MS setup at Octant is shown above. The CAD quantifies abundance, while the MS is responsible for CAD peak identification. Chromatographic separation is carried out with a 50 mm Waters ACQUITY HSS C18 column over a 4-minute method. Comprehensive method details can be found in the blog post’s GitHub repository.
Reaching the final CAD peak area-to-mass equation required developing a systematic method against a defined set of requirements. The method had to be (i) rapid enough to keep pace with HTChem synthesis throughput, (ii) reproducible across instrument runs, (iii) quantitative across structurally diverse analytes, (iv) accurate and specific enough to assign each chromatographic peak to a single chemical species, and (v) wide enough in dynamic range to quantify the range of yields encountered in HTChem. The first iteration of our method, run on a panel of analytical reference standards using common reverse-phase C18 chromatographic conditions, fell short on two fronts: individual calibration curves were nonlinear, and the response slope varied substantially from analyte to analyte.
The final diluent and mobile phase compositions selected for the method addressed the linearity issue and deviate from a typical reversed-phase C18 LC-MS configuration. Specifically, to mitigate the nonlinearity attributed to analyte solubility, MeOH is included in both the diluent and mobile phase at 20% v/v. At higher injection masses, the standard reverse-phase conditions could not keep lipophilic drug-like analytes fully solubilized at the moment of injection, compressing the dynamic range. A co-solvent of 20% MeOH restored linearity across our dynamic range of interest to adequately capture yield across the HTChem platform.
Apart from solvent effects (which were already addressed by the inverse gradient), the most reported and verified driver of variable CAD response is salt formation 13. When an analyte forms a salt with a mobile phase additive, the total mass of the CAD particle is the analyte plus its associated counterion. Our original mobile phase used formic acid, and our drug-like chemical matter contained variable numbers of basic nitrogen atoms that would protonate and ionically bond to formate counterions. Quinidine is a representative example with its two basic nitrogens (a tertiary amine, pKa 8.6, and a quinoline nitrogen, pKa 4.2). By readily forming a doubly protonated diformate salt under 0.1% formic acid-containing LC conditions, quinidine’s effective mass at the CAD detector is increased by 1.28x (Fig. 4). This can be addressed via “salt-mass correction,” in which CAD peak areas are adjusted on a per-analyte basis using well-characterized pKa values. However, our broader objective is the high-throughput characterization of diverse, novel chemical matter, and salt-mass correction is only as good as the pKa estimate. Existing pKa prediction tools such as Chemaxon and RDKit routinely disagree with experimental values (particularly for novel scaffolds) by amounts that would undermine salt-mass correction accuracy. For a known compound library, salt-mass correction is reasonable, but for the kind of chemical space HTChem is built to explore, it is not tractable.
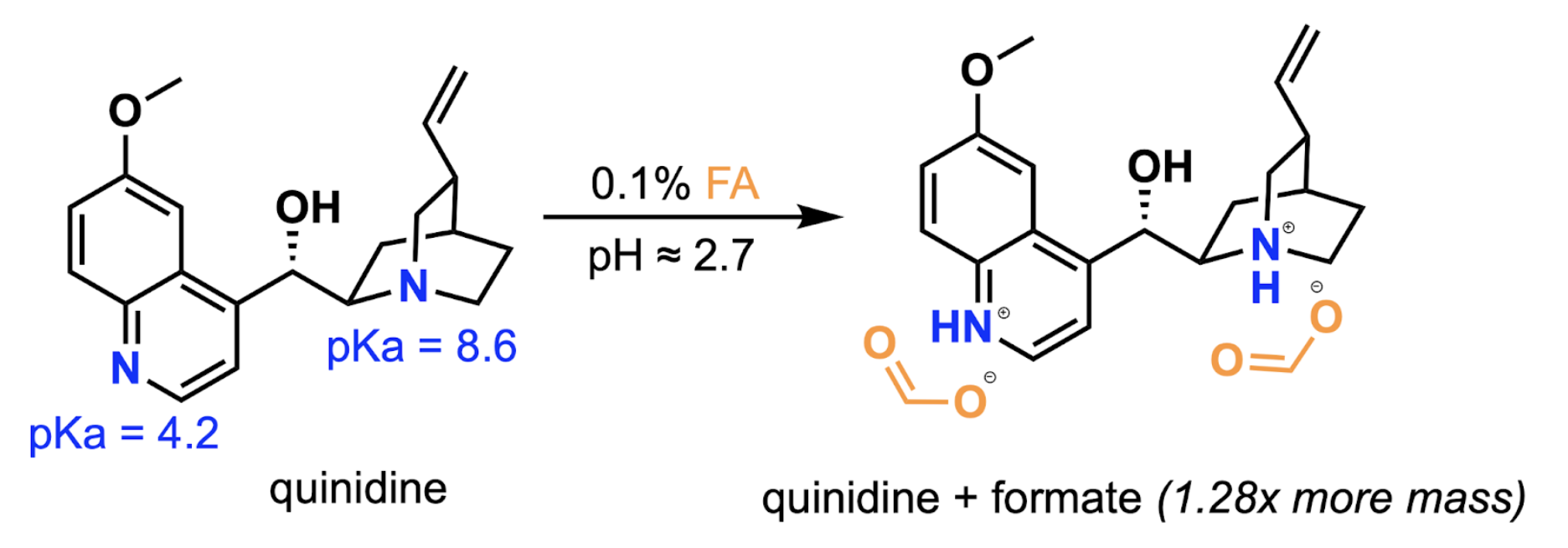
Figure 4: Quinidine’s nitrogen atoms are protonated at 0.1% formic acid (v/v), leading to formate salt complexes that inflate CAD peak area and lead to inconsistencies in response.
Rather than correcting for it, the cleaner solution was to remove formic acid from the mobile phase. With no formic acid, no formate salts form, and no per-analyte correction is needed. Predictably, due to formic acid’s role in controlling the pH and minimizing undesirable interactions with the stationary phase, the immediate consequence was a degradation of chromatographic peak shape. Through targeted reoptimization of the LC method—specifically, a 1.8 µm Waters ACQUITY UPLC HSS C18, a shallow gradient segment where most compounds elute, and pre-column compartment heating to 60 °C—chromatographic quality was recovered, and the corresponding tightening of inter-analyte CAD response confirmed that the salt-formation pathway had been a meaningful contributor to response variability.
To further improve linearity and reduce response variability, two parameters on the CAD itself were optimized: the power function value (PFV) and the CAD evaporator temperature (EvapT). A PFV of 1.15 produced the most robust linear response across diverse drug-like analytes and was implemented into the method. For the EvapT, it is known that higher temperatures decrease baseline noise by ensuring complete evaporation of mobile phase solvent microdroplets and semi-volatile trace impurities—improving signal-to-noise and lowering the achievable lower limit of quantification (LLOQ)—but push more intended analytes into the semi-volatile regime (where some fraction of the analyte mass evaporates with the mobile phase before reaching the detector) 13. By testing evaporator temperatures ranging from 20 °C to 95 °C against a reference panel consisting of molecules used in our ADMET assays, we identified 65 °C as the optimal compromise between noise reduction and preservation of non-volatile analyte behavior. However, some analytes of interest do behave as semi-volatile at 65 °C, but which molecules will behave this way cannot be confidently predicted in advance since volatility is governed by boiling point and enthalpy of vaporization, neither practical to measure nor reliably predicted. Molecular weight can serve as a general proxy for volatility, but in order to catch these cases of semi-volatility in a robust manner, we apply a routine QC step in which suspect analytes are reinjected with the EvapT lowered to its minimum (20 °C). A substantial increase in CAD peak area (beyond what would be expected from a noisier baseline) flags semi-volatile behavior, while a small change is consistent with non-volatile behavior. Although an EvapT of 20 °C produces a noisier baseline and therefore less precise yields, it provides more accurate yields than 65 °C for compounds that behave as semi-volatile at 65 °C. The cumulative effect of these systematic interventions—diluent and mobile phase optimization, formic acid removal, LC method recovery, PFV tuning, and EvapT optimization—was a UHPLC-CAD-MS method that produced near-uniform response across structurally diverse drug-like chemical matter. Ultimately, our protocol for determining yield utilizing CAD demonstrated robust validation, which in turn allows us to correct the EC50 of our HTChem-synthesized libraries.
4) Propagation of Standard Error for Corrected pEC50
Estimating the standard error of a corrected pEC50 value requires propagating the uncertainty associated with each component used to derive it. In our workflow, corrected pEC50 values are generated by adjusting an EC50 based upon a compound’s CAD-determined yield. The final standard error therefore includes three independent contributions: the DRC fitting error, the CAD peak area measurement error, and the between-compound variability in the universal CAD calibration slope. These contributions are combined according to Eq. 4 to approximate the standard error of a corrected pEC50.
\[\text{Eq. 4} \qquad SE_{\text{corrected pEC}_{50}} \approx \sqrt{SE(\text{measured pEC}_{50})^2 + \frac{CV(\text{Area})^2 + CV(\text{slope})^2}{\ln(10)^2}}\]
The first term of Eq. 4 is the DRC fitting error, \(SE(\text{measured pEC}_{50})\), which captures the precision with which the inflection point of the dose-response curve was determined and is independent of whether the underlying concentrations were nominal or yield-corrected.
The other sources of uncertainty arise from the CAD-based yield determination, including both the noise in the CAD peak area measurement and the variability in the universal calibration slope. The second term of Eq. 4 is \(CV(\text{Area})\), which characterizes the precision of the CAD peak area measurement at a given signal level. This measured coefficient of variation depends strongly on peak magnitude, with small peaks suffering disproportionately from baseline contributions while large peaks approach an asymptotic floor reflecting injector and detector variability. For any peak area measurement, \(CV(\text{Area})\) is computed from its own observed peak area (shown graphically in panel 2 of Fig. 5), yielding a per-compound noise estimate that scales appropriately with signal level. The third term of Eq. 4 is the between-compound spread in the universal CAD calibration slope, \(CV(\text{slope})\). The CAD “universal response” assumption enables concentration estimation without per-compound calibration, but it is inherently an approximation. We obtained a coefficient of variation of 31.74% by aggregating calibration slopes from a 300+ compound set spanning drug-like chemical space. This final term of Eq. 4 captures the expected compound-to-compound variation in CAD response when applying the universal slope to novel, uncharacterized compounds.
Since these three contributions are treated as independent, their variances are combined in quadrature. The DRC fitting error is already expressed in pEC50 units, whereas the CAD area and slope CVs are relative uncertainty estimates from linear-space quantities. Before these CV-based contributions can be combined with the DRC fitting variance, they must be converted to base-10 log units by dividing their squared values by \(\ln(10)^2\). With all terms expressed in pEC50 units squared, the square root of their sum gives the approximate standard error of the corrected pEC50.
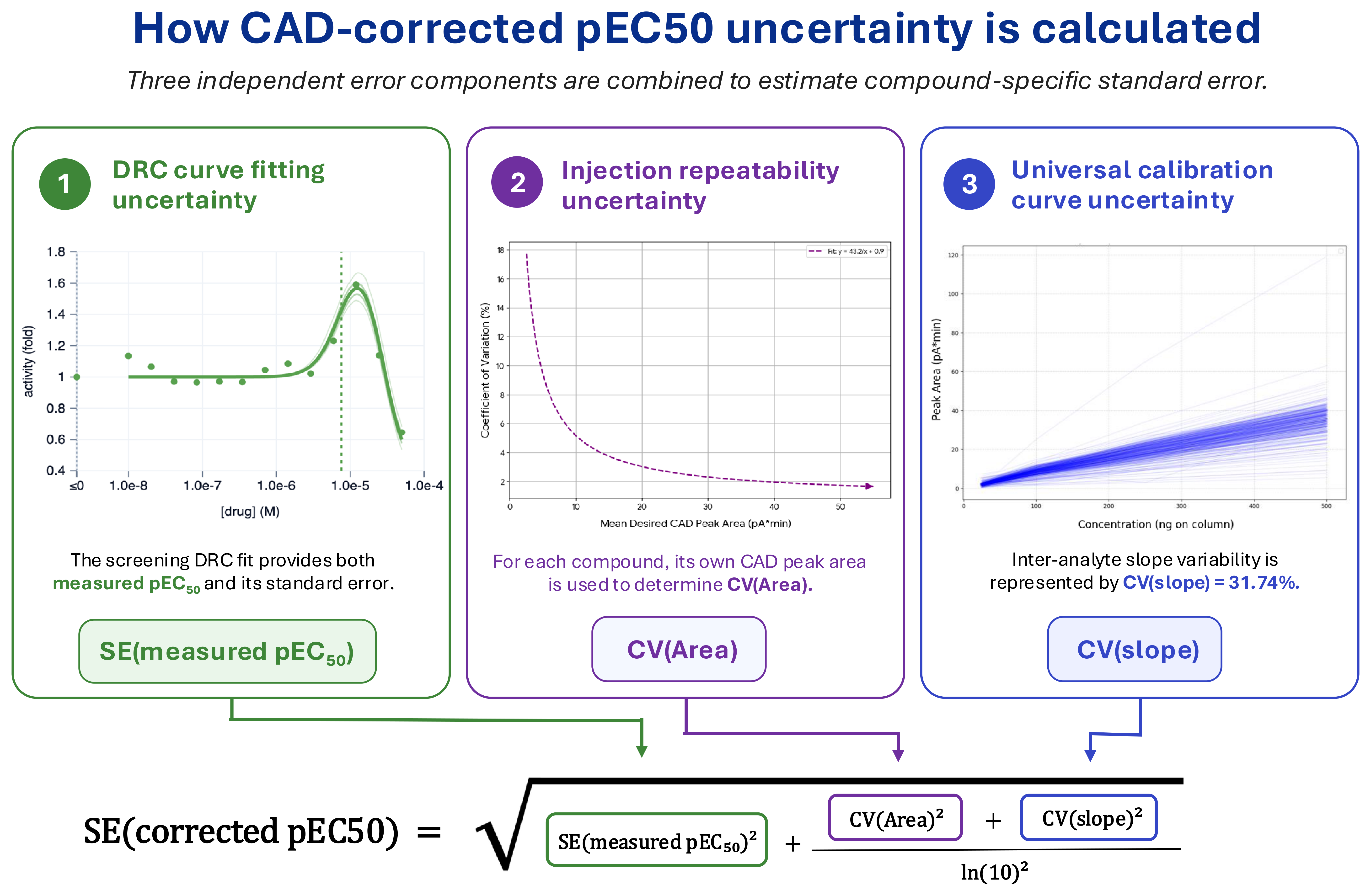
Figure 5: CAD-corrected pEC50 standard error is estimated by combining three independent contributions, which are the DRC fitting error (panel 1), the CAD peak area measurement error (panel 2), and between-compound variability in the universal CAD calibration slope (panel 3). The CAD-derived CV terms are converted to pEC50 units and combined with the DRC fitting error in quadrature to obtain a compound-specific standard error.
5) Validation of Yield-Based EC50 Correction
Next, to determine whether our standard-free absolute quantification UHPLC-CAD-MS method can robustly correct EC50s, we tested 20 compounds as synthesized crudes, “semi-purified” crudes, and purified standards. While the details distinguishing crudes from semi-purified crudes will be discussed in a future blog post, for the purposes of this post, semi-purification can be understood as a cleanup step that removes residual reaction inputs, whereas full purification isolates solely the intended product. Specifically, Octant’s method of semi-purification removes hydrophilic impurities from a crude reaction mixture, which, for amide couplings, constitute the majority of non-product reaction material due to the highly polar nature of the starting materials and reagents. Following DRC collection and CAD-based yield analysis of the synthesized compounds, we compared the pure pEC50 values to both the unadjusted and yield-adjusted pEC50s of the crudes (Fig. 6A,B). Applying this correction improved the agreement between the crude and pure pEC50, reducing the MAE (mean absolute error) from 0.447 (no correction) to 0.250 (with correction). Additionally, we see strong agreement (MAE = 0.168) between the corrected pEC50 of compounds in their crude and semi-pure forms (Fig. 6C). This observation indicates that the reagents and starting materials used in the synthesis of HTChem compounds, which are analytically-determined to be removed through semi-purification, do not have a large impact on the cell-based PXR-activation reporter assay. The filtering effect of the cell membrane is likely substantially reducing assay interference from polar impurities in this situation, but this protective effect would not apply to a biochemical assay. The method development for the synthesis itself warrants its own dedicated blog post, and will be discussed at a future date.
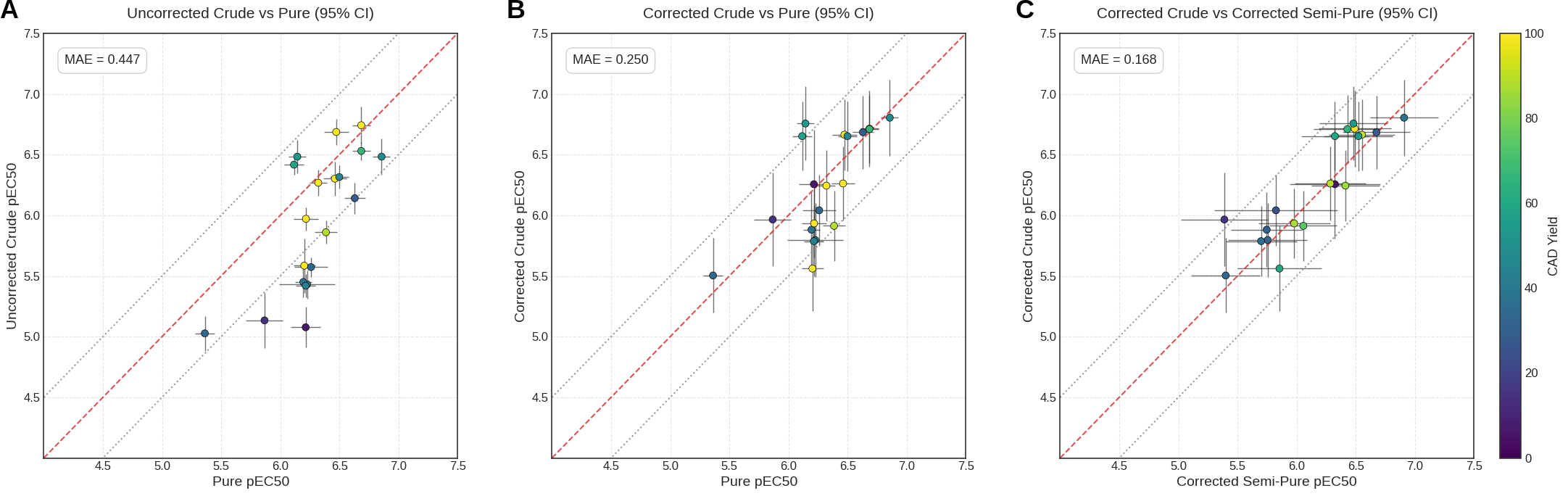
Figure 6: Twenty PXR agonists with existing pure-compound screening data were synthesized and screened as crude products. The same 20 compounds were then resynthesized as crudes, semi-purified, and screened again. This generated three matched screening datasets for each compound: pure, crude, and semi-pure. The comparison of uncorrected crude-to-pure pEC50 is shown on the left (A), the corrected crude-to-pure pEC50 comparison is shown in the middle (B), and the corrected crude-to-semi-pure pEC50 comparison is shown on the right (C). The diagonal red-dotted line of each plot represents complete agreement, and the gray dotted lines on either side represent ±0.5 pEC50 units. Crude and semi-pure sample concentrations were determined using CAD, and yield-adjusted pEC50 values were calculated accordingly. In panels A and B, points are colored by the CAD-determined crude product yield. In panel C, points are colored by the average yield of the corresponding crude and semi-pure samples. Error bars represent 95% confidence intervals. Plots were generated using Python.
These results allow us to return to the two original questions surrounding the use of D2B for model-training data generation. First, can the true concentration of a crude HTChem compound be determined at nanoscale? The CAD-MS yield-determination method provides a practical answer by enabling concentration correction without compound-specific standards. Second, is the observed assay activity driven by the intended product or by crude reaction impurities? The strong correlation between crude and semi-purified samples suggests that non-product reaction components—excess fragment, unreacted core, coupling reagents, and polar byproducts—do not substantially affect the assay readout. With these questions addressed, we can advance to larger-scale D2B experiments to explore the chemical space around PXR induction.
6) HTChem with CAD-MS to Explore PXR SAR
By bringing together HTChem, HTS, and CAD-MS, we are able to build, test, and characterize novel chemical libraries of interest. Octant’s chemistry platform facilitates the elucidation of structure-activity relationships with significantly higher resolution than is possible using commercial libraries. Figure 7 highlights an example of preliminary SAR derived from a 96-compound “semi-pure” HTChem library with CAD-corrected PXR assay data. Specifically, the initial compound (Compound A) is relatively inactive, with a pEC50 of 4.05 (89 µM). However, expanding the central piperazine to a seven-membered homopiperazine ring yields a 10-fold increase in potency (Compound B). Furthermore, introducing a 3-isopropyl group to the left-hand pyrimidine core (Compound C) enhances activity by nearly 100-fold. Interestingly, installing a polar methyl ester at this same position (Compound D) fails to elicit a significant increase in PXR activation, and an alternate pyrimidine substitution (Compound E) similarly provides no potency benefit. This example highlights how rich in activity cliffs PXR SAR can be, and reinforces the value of high-resolution SAR exploration with analogs from HTChem. A related series bearing an α-methyl substitution at the benzylic position (Compounds F-J) showed a similar SAR pattern. Consistent with the first series, we see an increase in pEC50 with lipophilic substitutions on the pyrimidine, while the polar methyl ester remains equipotent with the unsubstituted pyrimidine (Compound F). Moving forward, we will continue leveraging our HTChem platform to expand this SAR landscape. By tightly integrating our HTChem, assay, and structural biology efforts, we aim to deepen our understanding of PXR and apply these techniques to other Avoidome targets.
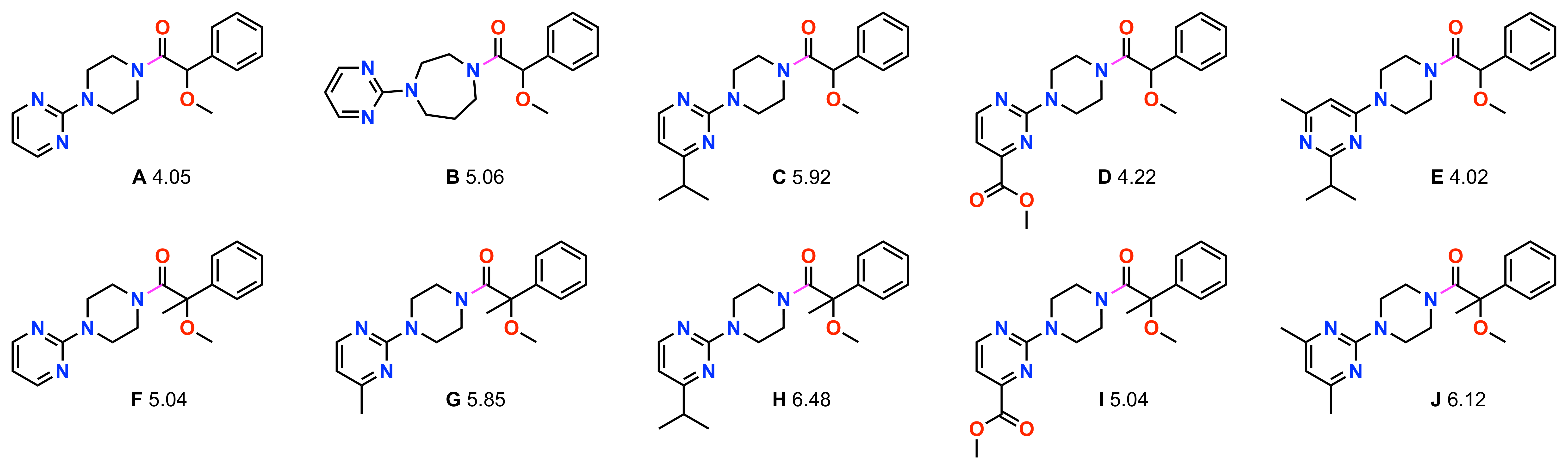
Figure 7: Ten related compounds from a 96-compound HTChem synthesis library are shown. The pink bond indicates the amide coupling linkage, and pEC50 values are shown underneath each compound.
From these HTChem SAR insights, the ability to characterize crude reaction yields, and our confidence that amide coupling impurities have limited impact on the PXR activation assay, we can proceed with additional HTChem library syntheses from two hit-derived carboxylic acid cores (Fig. 8). One particular core, OCNT-0002416, was designed from the α-methyl-substituted series (Compounds F-J in Fig. 7), which showed interesting SAR patterns that we wanted to explore further. The full HTChem libraries were screened at two concentrations—10 µM and 30 µM—to probe for activity while catching compounds with rollover activity (i.e., active at lower concentrations but losing activity at higher concentrations). Primary screen hits, as well as chemically-similar molecules to these hits (called “chemisimilars”), were selected for follow-up DRC collection to assess EC50. Specifically, 228 of the original 1,536 compounds per library had 9-point dose-response curves generated from 5 nM to 50 µM. With CAD-determined reaction yields for these 456 molecules, corrected pEC50s were calculated, and these data are presented in the interactive plots below (Fig. 9A-E).
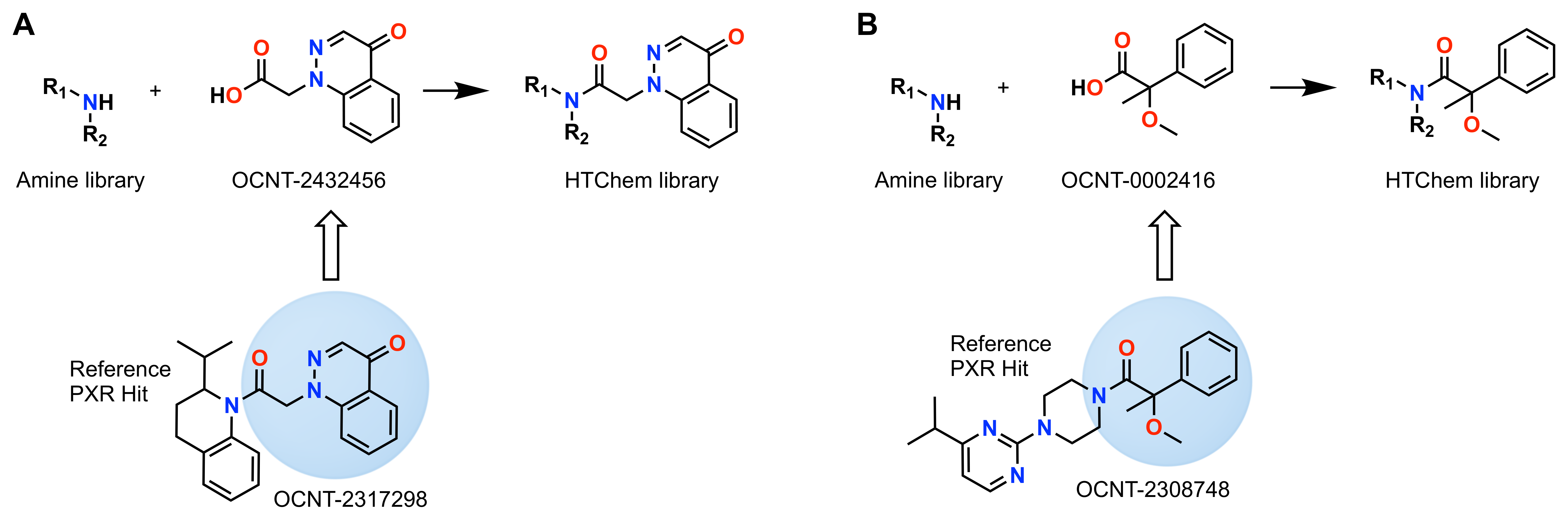
Figure 8: Two known PXR agonists were used to design carboxylic acid HTChem cores, which underwent amide coupling with a library of amine-bearing fragment molecules.
A. Reference hit OCNT-2317298 was used to design the OCNT-2432456 core, which was reacted with a collection of 1,536 diversity-selected amines.
B. Reference hit OCNT-2308748 was used to design the OCNT-0002416 core, which was reacted with a collection of 1,536 diversity-selected amines.
Figure 9A: Beeswarm plot presentation of the HTChem libraries, including the 96-compound microscale synthesis, color-coded by CAD-derived synthesis yield.
Figure 9B: OCNT-2432456 library — uncorrected vs. corrected pEC50, color-coded by chemical similarity to the reference hit.
Figure 9C: OCNT-0002416 library — uncorrected vs. corrected pEC50, color-coded by chemical similarity to the reference hit.
Figure 9D: pEC50 movement following yield-based correction for the 30 most potent hits from the OCNT-2432456 library, color-coded by chemical similarity to the reference hit.
Figure 9E: pEC50 movement following yield-based correction for the 30 most potent hits from the OCNT-0002416 library, color-coded by chemical similarity to the reference hit.
Figure 9: Two known PXR agonists were used to design carboxylic acid HTChem cores, which underwent amide coupling with a library of amine-bearing fragment molecules. Beeswarm plots of the HTChem libraries, including the 96-compound “semi-pure” microscale synthesis, is presented, with compounds color-coded based on CAD-derived synthesis yield (A). The two carboxylic acid-core HTChem libraries are plotted with respect to their uncorrected (x-axis) and corrected (y-axis) pEC50 values, with color coding based on chemical similarity to the reference hit from which the core was derived (B, C). The pEC50 movement following yield-based correction for the 30 most potent hits from the two carboxylic acid-core HTChem libraries is shown, with color coding based on chemical similarity to the reference hit (D, E).
From these HTChem syntheses, broad observations can be made, such as the OCNT-0002416 core library being more potent on average than the OCNT-2432456 core library, but also having more failed syntheses (Fig. 9A). Among the compounds from these libraries identified as primary screen hits or chemisimilars, we advanced both groups in parallel to DRC collection and CAD quantification. In contrast to primary screen hits, chemisimilar compounds are not active in primary screens, possibly due to failed synthesis, insufficient product yield, or a true activity cliff. Using our yield quantification capabilities, we can identify compounds with insufficient yield that would otherwise have led to false-negative data. This consideration highlights the importance of integrating CAD with HTChem for model-training purposes, where low-yielding or failed syntheses could otherwise be misclassified as true biological inactivity.
Together, the three HTChem libraries, including the 96 “semi-purified” crudes, expand the publicly-available PXR screening dataset and are intended to facilitate further exploration of PXR SAR across related scaffolds. All raw data for the libraries and experiments described in this post—including CAD peak areas, CAD-derived yields, pEC50 values, and yield-corrected pEC50 calculations with error estimates and confidence intervals—are available in the accompanying GitHub repository.
The crude and semi-pure HTChem library data released with this post are also available on HuggingFace at openadmet/pxr-challenge-train-test. They can be loaded with the datasets library:
from datasets import load_dataset
# Crude HTChem libraries (454 novel EC50s)
ds_crudes = load_dataset("openadmet/pxr-challenge-train-test", "crudes_htchem")
train_crudes = ds_crudes["train"]
# Semi-pure HTChem library (96-compound microscale synthesis)
ds_semi_pure = load_dataset("openadmet/pxr-challenge-train-test", "semi_pure_htchem")
train_semi_pure = ds_semi_pure["train"]For the crude libraries, key columns include SMILES, OCNT_ID, Corrected Crude pEC50 (log), Corrected Crude pEC50 ±95% CI (log), Crude Product Yield (%), and Crude Peak Area (pA*min). The semi-pure dataset shares the same schema, with yield and pEC50 values reflecting the semi-purified sample format.
7) Conclusion
The methodologies we have described address a fundamental tension in high-throughput data generation: the trade-off between scale and fidelity. By integrating standard-free UHPLC-CAD-MS quantification with cell-based screening, we can estimate compound-specific reaction yields at the nanoscale, correct nominal D2B assay concentrations, and convert crude HTChem screening results into quantitative pEC50 values.
This yield-correction step is what allows HTChem data to move beyond hit finding. Instead of treating crude library screening as a preliminary activity signal that requires downstream resynthesis before it can be trusted, CAD-MS enables each compound’s apparent EC50 to be interpreted in the context of how much product was actually formed. This capability is crucial for generating model-training data, in which HTChem compounds with low reaction yields or failed syntheses could otherwise be underestimated in potency or misclassified as inactive.
Applied to PXR, this workflow generated new open datasets spanning crude, semi-purified, and purified compound formats, as well as larger HTChem libraries designed around known PXR activators. More broadly, the same approach can be used to rapidly generate quantitative SAR data around other Avoidome targets. We are excited to make this expanded PXR dataset available to the OpenADMET community and invite feedback through the OpenADMET Discord or by email.
8) Acknowledgments
The authors would like to acknowledge experimental data, analysis, and scientific input contributed by Ayesha Ghazali, Bryan Jiang, Galen Correy, Hugo MacDermott-Opeskin, Junyu Deng, Marco Falcioni, Naomi Handly, Nathaniel Luis, Patrick Walters, Robert Warneford-Thomson, Sam Sabaat, Scott Simpkins, Steven Edgar, and Theodore Tarver.
This work was supported by funding from ARPA-H and the Astera Foundation.